Mapped Reads (BAM)¶
Introduction¶
When data that comes out of a sequencer is mapped to a reference genome, the result is a set of mapped reads that can be visualized as a pileup. If this data is provided in an indexed BAM file, it can be visualized in Resgen.
Tags¶
filetype:bam datatype:alignments
Data Preparation¶
There are lots of resources describing how to map sequencing data to a reference genome. A few of the commonly used tools are:
- Bowtie - Another short read mapper
- BWA - For short read mapping
- Minimap2 - By the author of BWA but for long read mapping
A mapper will take an input file (typically FASTQ or FAST5) containing sequencing reads, map it to a reference genome and generate a SAM or BAM file. This resulting BAM file can then be sorted and indexed to prepare it for visualization:
samtools sort my.bam -o my.sorted.bam
samtools index my.sorted.bam
The result of this step will be two files: my.sorted.bam containing the sorted mapped reads and its index: my.sorted.bam.bai. For a much more comprehensive overview of read alignment and what comes after, take a look at Erik Garrison's Alignment and Variant Calling Tutorial.
Loading into Resgen¶
Files¶
To load BAM files into resgen, we first need to create a project. Then click on "Add Dataset".
URLs¶
BAM files can also be added as URLs. To do this we will click on the "Add URL" tab of the "Add Dataset" dialog and then paste in the URL of the BAM file. In this example we will use the following URL:
https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_HiSeq_HG002_Homogeneity-10953946/NHGRI_Illumina300X_AJtrio_novoalign_bams/HG002.hs37d5.60x.1.bam
Entering a URL with a .bam extension will make the "Index URL" field appear. This field will be populated with the expected location of the .bai index file: <filaname>.bam.bai

Click on "Submit" to add the file to the project. You should then be able to drag the eye icon onto the viewer as described in the Viewing Data section.
Associating with an assembly¶
BAM files contain reads mapped to a reference composed of multiple sequences (or contigs). Because HiGlass displays multi-contig data end-to-end as if individual contigs were concatenated, it needs to know which order arrange them in. This can be done by associating an assembly with the BAM dataset.
An assembly, as far as Resgen and HiGlass are concerned is a list of chromosome sizes arranged in a particular order and tagged with the assembly: tag. When loading a BAM file, Resgen will look for a chromsizes dataset with the same assembly: tag as the BAM dataset in the current project. If it doesn't find one, it will try to naturally order the chromosomes listed in the BAM file.
For this example, we will add this b37 chromsizes file and tag it as assembly:b37

We can now add the chromosome sizes and BAM file to view the data. Note that when the datasets are first added there is no zoom level set so HiGlass displays the entire genome. When the BAM file is added, we need to zoom in to see its contents.
Available options¶
As with all other tags, options are set using the track's config menu. See the "Configuring tracks" section of the "Viewing data" tutorial for an overview.
Group by¶
 Reads grouped by the HP tag
Reads grouped by the HP tag
Outline reads on hover¶
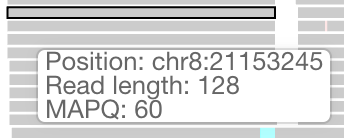 Read outlined above tooltip
Read outlined above tooltip